
Mitochondrial Trafficking: A Two-Way Street
- Feb 24, 2022
- 5 min read
Opposing microtubule motors, such as the plus-end-directed kinesin and the minus-end-directed dynein, must work in tandem to actively transport cargo within the cell. Although some cargos are unidirectionally transported by individual motor proteins, a significant portion of cellular cargo is transported in a bidirectional manner by two distinct motor proteins. In the case of bidirectional transport, the opposing motors must precisely coordinate their movements to guarantee efficient transport and localization. Mitochondrial transport is a typical example of bidirectional transport, as organelles are shuttled along the microtubule cytoskeleton by the kinesin-1 (KIF5) family of motor proteins and the dynein-dynactin complex to meet local energy requirements and maintain cellular homeostasis. Bidirectional mitochondrial transport is critical in elongated cells, such as neurons, and transport failures can result in detrimental neurodegenerative disorders like Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. To date, the exact mechanisms behind opposing motor coordination in mitochondrial transport were unclear.
Under the mentorship of Dr. Erika Holzbaur and Dr. Thomas Jongens, Adam Fenton, a CAMB Genetics and Epigenetics student, proposed a novel model to explain the motor coordination behind mitochondrial trafficking, as reported in their recent Nature Communications article. Previous work identified the TRAK/Milton family of proteins as the mitochondrial motor adaptors that connect Miro, the mitochondrial outer membrane protein, to kinesin-1 and the dynein-dynactin complex. Specifically, others have postulated that TRAK1 serves as the motor adaptor that interacts with kinesin-1 and the dynein-dynactin complex in axonal mitochondrial transport, whereas TRAK2 serves as the motor adapter that primarily interacts with the dynein-dynactin complex in dendritic mitochondrial transport. However, recent studies have mapped both kinesin-1 and dynein-dynactin binding interfaces to TRAK2, raising the paradox/question of whether TRAK2 can simultaneously coordinate these two opposing motors. Interestingly, neuronal knockdown of kinesin-1, dynein, or dynactin individually is sufficient to inhibit mitochondrial transport, suggesting the co-dependent relationship. With these previous observations in mind, Adam Fenton and his mentors aimed to develop a system that allowed them to observe the exact molecular interactions between TRAK proteins and the two opposing motor proteins in question, kinesin-1 and the dynein-dynactin complex.
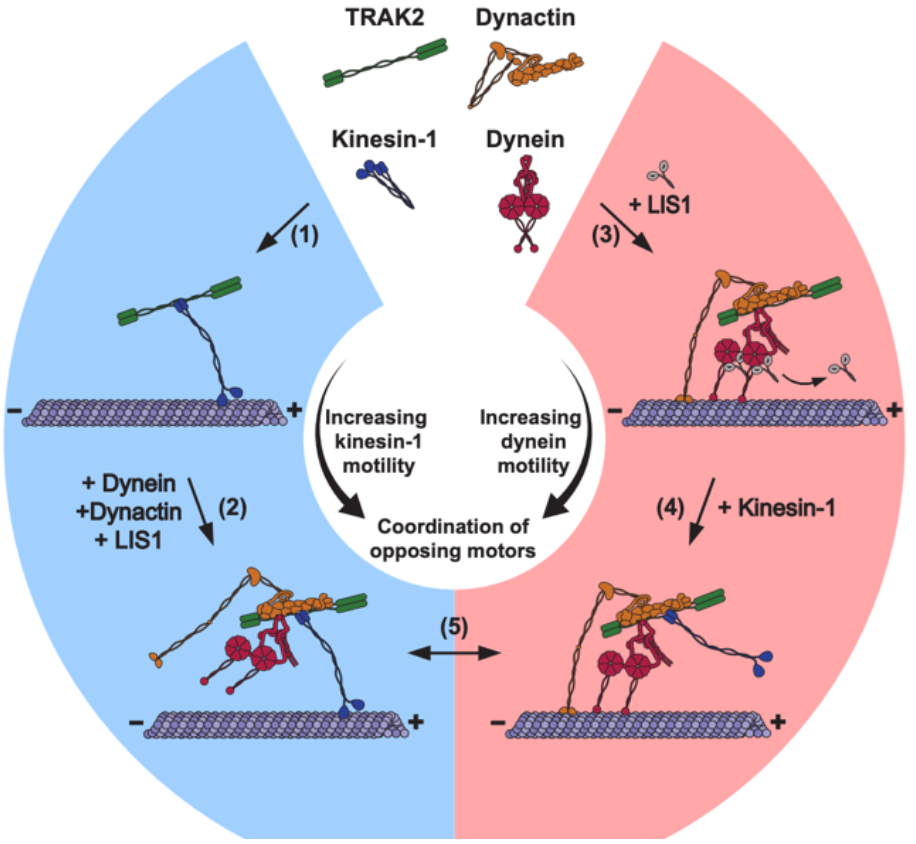
To examine the functional effects of TRAK2 interaction on kinesin-1 and dynein, the authors devised an in vitro single-molecule approach by Halo-tagging TRAK2 in COS-7 cells labeled with a tetramethylrhodamine (TMR)-HaloTag ligand. Next, Fenton and colleagues used total internal reflection fluorescence (TIRF) microscopy to observe the motility of the TRAK2-motor complex in the generated cell lysate. This innovative single-molecule approach allowed the authors to identify three distinct motility patterns: the TRAK2 motor complex exhibited unidirectiona
l movement towards the plus-end and minus-end, as well as shorter back-and-forth movements. Despite this variety, the authors recognized that the TRAK2-motor complex favored processive, unidirectional movement towards the microtubule plus-end compared to the short, infrequent, and varied movement towards the microtubule minus-end. Noticing this preference, Adam Fenton hypothesized that TRAK2 was responsible for inducing processive, plus-end-directed transport through kinesin-1 activation.
Aware that minus-end motility was nearly absent in this Halo-tagged TRAK2 motor complex, the authors noted that they were missing a critical component of this motor complex required for dynein activation. The most obvious candidate for induction of minus-end motility was LIS1, a protein found to activate processive dynein motility in previous studies. Fenton et al. exogenously expressed HA-tagged LIS1 in COS-7 cells with Halo-tagged TRAK2 and used TIRF microscopy to determine what effect LIS1 has on the complex’s motility. Adding LIS1 to the Halo-tagged TRAK2-motor complex resulted in increased dynein-mediated processivity and velocity towards the minus-end and no apparent effects on plus-ended movement, suggesting that the TRAK2-motor complex and LIS1 work in conjunction to activate the dynein motor protein.
Fenton and colleagues then sought to determine if the TRAK2 CC1-Box, a domain that is conserved amongst dynein activators, is required to activate dynein-dynactin in the TRAK2-motor complex. First, they introduced point mutations within the Halo-tagged TRAK2 and introduced HA-LIS1 back into the system. TIRF microscopy of the co-expressed HA-LIS1 and Halo-TRAK2 in COS-7 cells revealed that mutations within the TRAK2 CC1-Box impaired minus-end motility, suggesting failed activation of the dynein-dynactin complex. Conversely, the plus-end motility remains intact with the point mutations. Furthermore, immunoprecipitation of the mutated Halo-TRAK2 protein, the Myc-tagged kinesin protein KIF5B, and the endogenous dynein heavy chain (DHC) confirmed that the CC1-Box of the TRAK2 protein is ultimately responsible for facilitating its interaction with the dynein-dynactin complex rather than kinesin-1. Taken all together, the authors reasoned that the presence of LIS1 and the TRAK2 CC1-Box domain specifically activates the dynein-dynactin complex.
Fenton then decided to explore further the tandem nature of kinesin-1 and dynein-dynactin in the TRAK2-motor complex. To accomplish this, siRNAs were used against kinesin-1 and dynein in COS-7 cells expressing Halo-TRAK2 alone or co-expressing with HA-LIS1. Interestingly, the authors noticed that siRNA knockdown of kinesin-1, irrespective of LIS1, resulted in the near inhibition of plus-ended motility in TRAK2 transport, subsequently confirming kinesin-1 as the primary motor responsible for plus-ended motility in TRAK2 transport. Surprisingly, Fenton et al. also noticed that siRNA knockdown of dynein resulted in a milder plus-ended motility decrease than the kinesin-1 knockdown. Based on these results, Fenton and colleagues concluded that the initiation of kinesin-dependent TRAK2 motility is amplified by the presence of dynein and dynactin, all independent of dynein activation by LIS1.
Thus far, the field’s current understanding of bidirectional transport and the exact role of TRAK proteins were unclear. Previous studies proposed similar but separate functions for the TRAK proteins: TRAK1 was the kinesin-1 motor adaptor responsible for axonal mitochondrial transport, while TRAK2 was thought to be the dynein-dynactin motor adaptor responsible for dendritic mitochondrial transport. However, Adam Fenton’s graduate research blows the field’s current understanding entirely out of the water. Functional studies of single TRAK2 motor complexes allowed Adam Fenton to conclude that TRAK2 interacts with both dynein-dynactin and kinesin-1, but preferentially activates kinesin-1 with or without the presence of dynein-dynactin. Additionally, they found that the presence of LIS1 enhanced the frequency, run length, and transport velocity towards the microtubule minus-end. Furthermore, the LIS1 enhancement of minus-ended motility depended on the TRAK2 CC1-Box domain, which they hypothesized to facilitate interactions with the dynein light intermediate chain. Overall, Fenton et al. proposed a novel model of mitochondrial transport: multi-motored TRAK2 complexes exhibit interdependence and loss or inhibition would result in diminished mitochondrial transport.
When asked about what inspires his work, Adam noted, “I’m interested in understanding how mitochondrial homeostasis is maintained in neurons. Mitochondrial transport is an extremely important process in neurons due to their extreme size and shape. We know that TRAK proteins play essential roles as adaptors for both kinesin and dynein on mitochondria during transport, but it was unclear how TRAK proteins could scaffold two motors that move in opposite directions.” In the future, Adam Fenton hopes to build a 3D model of the TRAK motor complexes to better understand mitochondrial transport. To accomplish this goal, he is collaborating
with Dr. Yi-Wei Chang of the Biochemistry and Biophysics department to visualize this cocomplex in-vitro and in neurons with cryo-electron tomography.
Reference:
Fenton, A. R., Jongens, T. A. & Holzbaur, E. L. F. Mitochondrial adaptor TRAK2 activates and functionally links opposing kinesin and dynein motors. Nat. Commun. 12, 4578 (2021)
Comments